
Enfermedad de Huntington
¿QUÉ ES LA ENFERMEDAD DE HUNTINGTON?
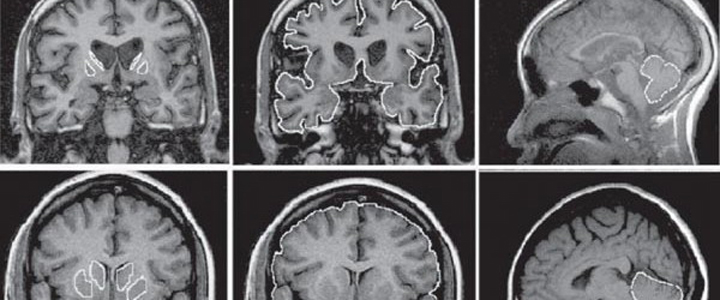
La enfermedad de Huntington es un trastorno neurodegenerativo en el cual algunas partes del sistema nervioso central pierden su funcionalidad a causa de la muerte de las neuronas que allí se ubican.
¿CUÁL ES SU CAUSA?
Es una de las enfermedades hereditarias producidas por la alteración en la secuencia de un gen que codifica para una de las proteínas que median funciones en el cerebro. Su herencia es autosómica dominante, de modo tal que cada hijo de un padre o madre afectado tiene un 50% de riesgo de heredar la alteración genética causante de la Enfermedad de Huntington. Esta enfermedad genética tiene la particularidad de producirse por un defecto molecular caracterizado por un número excesivo de repeticiones en una parte de la secuencia del gen Huntingtina. El número de estas repeticiones puede modificarse generación tras generación (aumentando o disminuyendo). Cuanto mayor sea el número de repeticiones, mayor será la posibilidad de presentar síntomas a una edad más temprana.
En general suele comenzar la sintomatología entre los 40 a 50 años de edad (el hecho de ser heredada de alguno de los padres no dictamina que la sintomatología se presente desde el nacimiento de la persona). Sin embargo, algunos pacientes pueden empezar a una edad menor (ver arriba modificación de número de repeticiones), aunque representan el menor número de casos.
¿CUÁLES SON LOS SÍNTOMAS?
En las etapas iniciales de la enfermedad, los síntomas incluyen cambios sutiles de coordinación, tal vez algunos movimientos involuntarios menores, dificultad para pensar en problemas y, frecuentemente, ánimo deprimido o irritable. En la etapa intermediaria, la corea (movimiento involuntario de los brazos y piernas) ya se torna prominente y la dificultad para llevar a cabo movimientos voluntarios es más evidente, acompañada de un empeoramiento de la disartria (alteraciones en el habla) y de la disfagia (dificultad para tragar). A medida que las deficiencias cognitivas aumentan, el paciente estará incapacitado de mantener una responsabilidad de trabajo o de llevar a cabo labores del hogar. Los pacientes en etapas tardías de la enfermedad, pueden presentar corea severa, pero más frecuentemente se encuentran rígidos y bradicinéticos (con movimientos lentos). Por lo general no pueden comunicarse, están confinados a una cama con una demencia más global, pero aún así retienen un grado significativo de comprensión verbal. Las alteraciones psiquiátricas se pueden presentar en cualquier etapa de la enfermedad, inclusive varios años antes del desarrollo de los síntomas motores.
¿CÓMO SE DIAGNOSTICA?
El especialista realizará un interrogatorio para ver antecedentes familiares y síntomas del paciente en cuestión. Luego podrá solicitar un estudio molecular que buscará cuantificar el número de repeticiones en la región afectada de la secuencia del gen Huntingtina con el objetivo de establecer si está anormalmente aumentado y será causante de la enfermedad.
ESTUDIO GENÉTICO PARA LA
ENFERMEDAD DE HUNTINGTON
Los estudios genéticos brindan una información que en algunas circunstancias permiten comprender mejor el pronóstico evolutivo de la enfermedad y los riesgos de recurrencia en la familia, siendo por lo tanto una herramienta diagnóstica sobre la que podrá consultar con su médico de cabecera.
Al igual que con otras enfermedades hereditarias, nunca debe solicitarse un estudio que pueda revelar un diagnóstico de una alteración genética responsable del desarrollo de una enfermedad que se manifiesta sintomáticamente en la edad adulta a un niño que aún no presenta síntomas; es decir, asintomático. Realizar un estudio genético a un menor de edad, asintomático, es considerado una falta ética que resulta en la violación de la autonomía inherente a todos los seres humanos.
Fuentes consultadas: www.neurogenetica.info – http://espanol.ninds.nih.gov/
Dr. Marcelo Kauffman @Marcelokauffman






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}